Hi! I'm Wiona, a computational biologist researching language models for immunology.
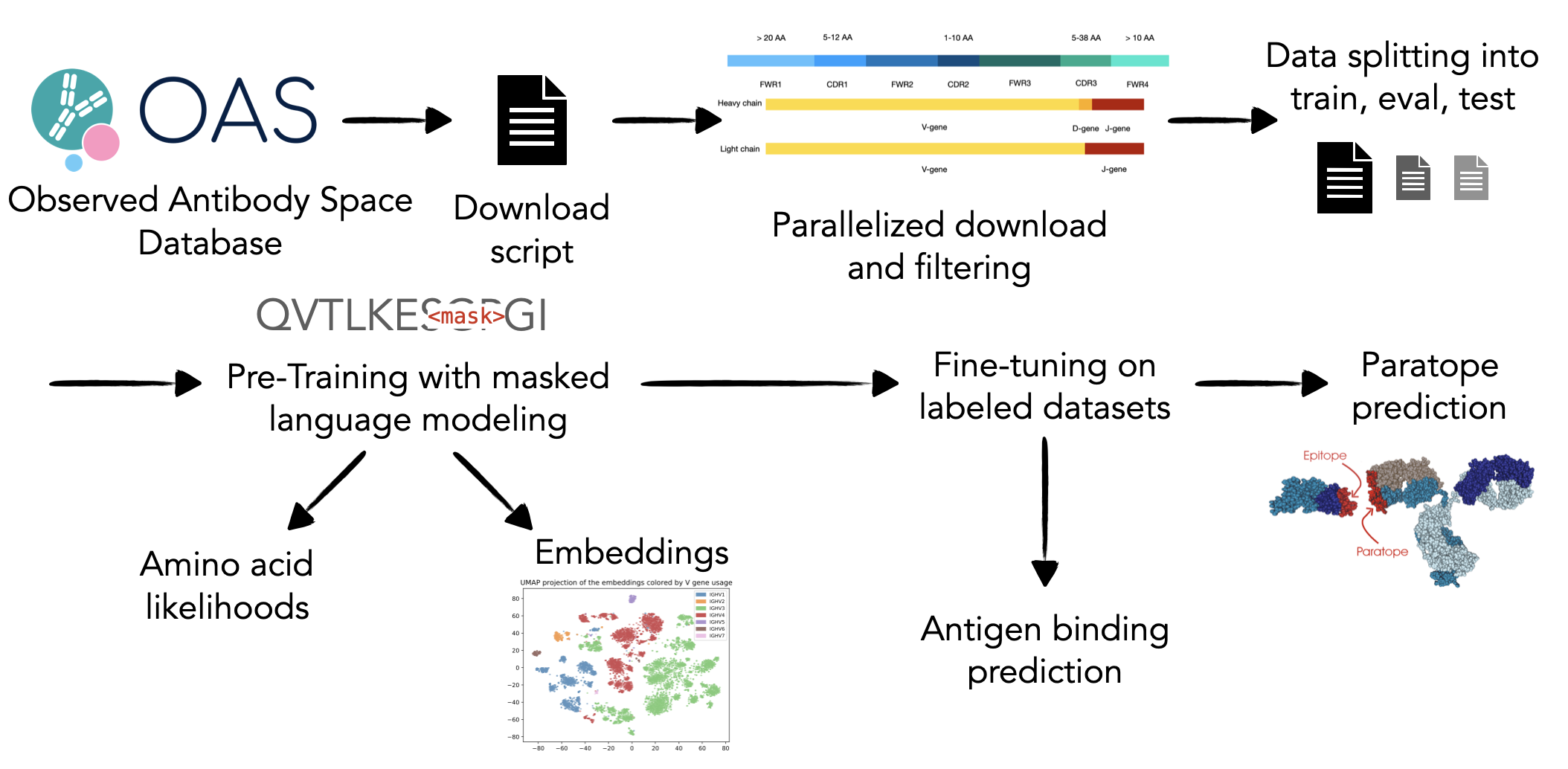
Antibody language models (LMs) trained on immune receptor sequences have been applied to diverse immunological tasks such as humanization and prediction of antigen specificity. While promising, these models are often trained on datasets with limited donor diversity, raising concerns that biases in the training data may hinder their generalizability. To quantify the impact of biased training data, we introduce an open-source processing pipeline for the 2.4 billion unpaired antibody sequences in the Observed Antibody Space (OAS) database, enabling customizable filtering and balanced sampling by donor, species, chain type and other metadata. Analysis of OAS revealed that 13 individuals contribute over 70% of human antibody sequences. Using our pipeline, we trained 17 RoBERTa antibody LMs on datasets of different compositions. Models failed to generalize across chain types and showed limited transfer between human and mouse repertoires. Both individual- and batch-specific effects influenced model performance, and expanding donor diversity did not improve generalization to unseen individuals from unseen publications.
Learn more
Antibodies are one of the most important proteins of the adaptive immune
system. They specifically recognize and bind to antigens to block their
function, remove them from the bloodstream, or mark pathogens for immune
cells. To recognize a variety of antigens, antibodies exhibit an immense
genetic diversity of over 1013 unique sequences in humans
and evolve within each organism in response to antigen exposure.
Understanding the mechanisms behind the generation of antibody
sequences is fundamental to illuminating the function of our immune
system and leveraging it for the treatment of disease.
For my Master's thesis I trained antibody language models on data
from the Observed Antibody Space (OAS) database and compared the performance
when training data from different species is used.

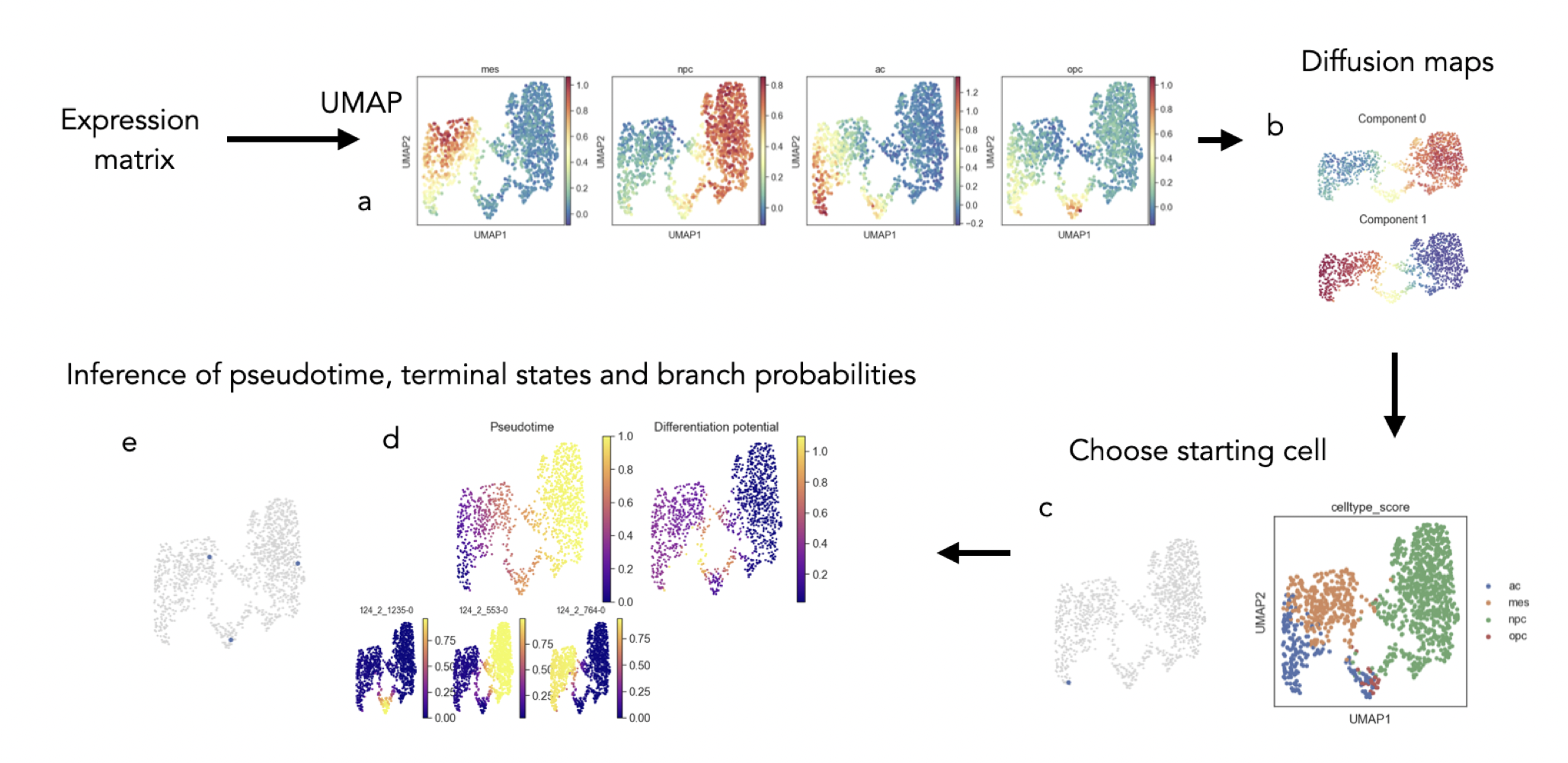
Glioblastoma is a hard-to-treat malignant brain tumor characterized
by high cellular diversity. Previous research has identified expression
meta-modules that define cellular states within Glioblastoma and cells
that appear to be in transition between these states. In this project,
I tested whether trajectory inference methods, developed to analyze
cell differentiation processes, could be utilized to identify trajectories
connecting these cell states. While some reservations remain about
the validity of this approach, the tested methods successfully
identified the expected trajectories and revealed gene expression
changes along them. These findings suggest that trajectory inference
techniques might offer valuable insights into Glioblastoma’s cellular
heterogeneity.

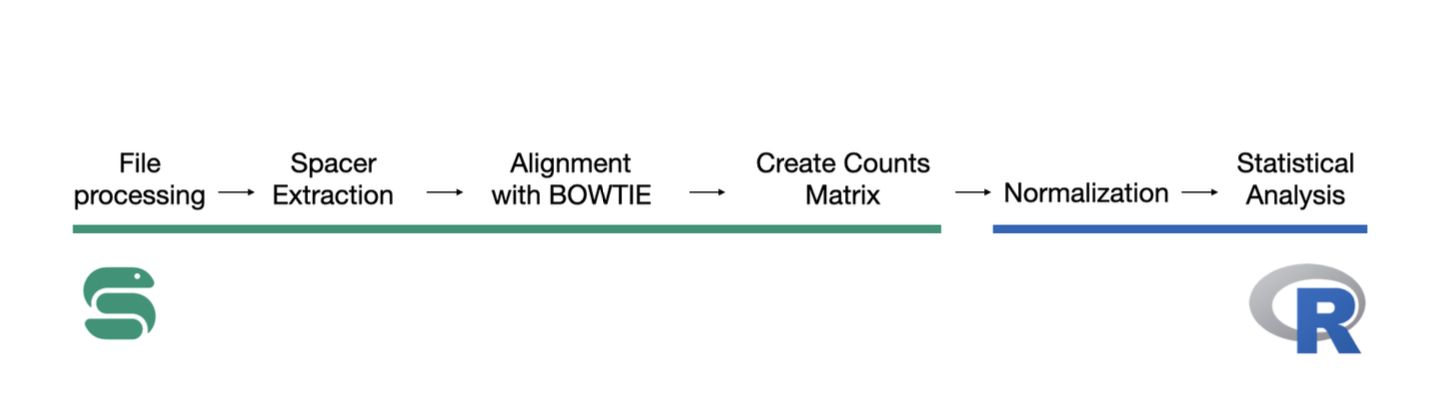
Record-seq is a recently developed technology that enables bacterial cells to record their own transcriptional
activity into DNA and preserve it over time. Record-seq readout consists of deep sequencing and data interpretation
using a dedicated computational analysis pipeline. In this project made improvements
to and conducted experiments with the Record-seq data analysis pipeline.
First, I worked on the primary analysis pipeline, augmenting its
capability to detect recorded sequences from reads using updated approximate
string matching methods. Next, I tested several methods for normalizing counts
distributions during secondary analysis to apply differential expression analysis
tools developed for RNA sequencing. Two new normalization methods, namely plasmid
normalization, and percentile normalization were implemented.